Update 6/26/26: “Sickle cell breakthrough: World’s first CRISPR therapy cures Louisiana man’s lifelong disease”
Goal of this exercise: Analyze the first study that used the CRISPR-Cas9 system (CASGEVY) as a treatment for Sickle cell disease (Frangoul et al. 2021*), and compare this with an alternate treatment (Risto-cel) that utilizes base editing (Gupta et al. 2026**). Part 1 below provides background information on the disease, outlines the therapeutic protocol and summarizes the molecular mechanism of CRISPR/Cas 9 and base editing treatments.
Note: The original sickle cell case involves genetic testing on family members in different case scenarios. The mobile version of the original case also uses NCBI tools to explore of the nature of the sickle cell mutation.
______________________________________
*Full text available via the above link
**You may be able to access the full text via your educational institution.
Part 1: Overview of CRISPR-Cas9 and base editing treatments for Sickle cell disease (this page).
Part 2: CRISPR-Cas9 exercise. Use Case It v705 to locate and cut the BCL11A erythroid-specific enhancer, then use BLAST and the Genome Data Viewer to visualize the enhancer.
Part 3: Base editing exercise. Use Case It v705 to locate and edit BCL11A binding sites on the HBG1 and HBG2 promoters, then use BLAST and the Genome Data Viewer to identify and visualize binding sites in relation to gene location.
Organization of Part 1
(1) Overview of Sickle cell disease
(2) Therapeutic concept of CRISPR-Cas9 treatment and base editing treatments
(3) Molecular mechanism of CRISPR-Cas9 treatment and summary
(4) Molecular mechanism of base editing treatment and summary
Overview of Sickle cell disease
Sickle cell disease (SCD) arises from a single point mutation in the β-gene of the HBB locus, encoding the β-globin subunit of adult hemoglobin (HbA). This mutation substitutes glutamic acid with valine, producing sickle hemoglobin (HbS). Fetal hemoglobin (HbF) does not contain beta-globin, and hence is free of the mutation.
{kind=link}
- Click the video screen below for an overview of the disease including its evolutionary relationship with malaria.
- Click this link to see a another video animation showing how HbS polymerizes in deoxygenated conditions, deforming red blood cells into rigid, crescent shapes, causing anemia, vaso-occlusive crisis, and organ damage.
Therapeutic concept
The mutation that causes sickle-cell disease occurs only in the beta-globin chains of adult hemoglobin. Enhancing the production of fetal hemoglobin via CRISPR or base editing treatments can mitigate effects of sickle cell disease.
Fetal hemoglobin (HbF) consists of two alpha-globin chains and two gamma-globin chains (α₂γ₂). Fetal hemoglobin has a much higher affinity for oxygen than adult hemoglobin (HbA), which consists of two alpha-globin chains and two beta-globin chains (α₂β₂). Click here for a video showing the location and composition of hemoglobin gene clusters in relation to the different types of hemoglobin produced during a person’s lifetime.
Before birth, most of the body’s hemoglobin is the (α₂γ₂) type. Shortly before or after birth, the body naturally transitions from producing gamma chains to beta chains, switching to adult hemoglobin. This structural difference allows the developing fetus to effectively obtain oxygen from the mother’s bloodstream across the placenta. This is critical because the fetus’s own lungs are not yet functional for gas exchange. By about 6 months of age, most of a baby’s hemoglobin is adult hemoglobin.
If a child receives the mutation from both parents, then all of the beta-globin chains will have the mutation, resulting in sickle-cell disease (homozygous recessive). If the child receives the mutation from one parent but the normal gene from the other parent, then they are heterozygous and would be asymptomatic for the disease. This is because a relatively small percentage of the hemoglobin has the mutation in both beta-globin chains, not enough to cause symptoms. Hemoglobin molecules with the mutation in only one of the two beta-globin chains function normally.
CASGEVY treatment: Fetal hemoglobin expression is normally repressed after birth by the transcription factor BCL11A, which binds to enhancer regions controlling γ-globin gene expression and silences them. CRISPR treatment targets the erythroid-specific enhancer for BCL11A and silences it, resulting in the continued production of fetal hemoglobin. Since it only targets this specific enhancer, CRISPR treatment does not adversely affect other critically important functions of BCL11A.
Risto-cel treatment: This base editing strategy differs from CASGEVY by targeting the promoter regions of the genes that produce gamma globin (HBG1 and HBG2). Normally, BCL11A binds to these promoter regions and suppresses them during the switch from fetal to adult hemoglobin. Base editing to switch a single base pair prevents BCL11A from binding to the promoter sites, allowing enough fetal hemoglobin production to prevent sickling in patients. It also differs from CASGEVY in that only one strand of the DNA molecule is cut (nicked) during the editing process, potentially reducing the risk of unintended genetic alterations. Unlike CASGEVY, risto-cel has not yet been approved for clinical use (as of July 2026) but clinical trials have shown promising results.
| Feature | CASGEVY | risto-cell |
|---|---|---|
| Mechanism | Silences the BCL11A repressor gene, allowing the production of fetal hemoglobin in adults. | Inhibits BCL11A from binding directly to the HBG promoters without modifying overall BCL11A levels. |
| Binding Site | Erythroid-specific enhancer region of the BCL11A gene. | HBG1 and HBG2 promoter regions. |
| Cas9 cut type | Double-strand break (DSB). | Single-strand nick (it uses a Cas9 nickase). |
| Pros | First FDA-approved CRISPR/Cas9 therapy. • Eliminates the need for recurrent blood transfusions. • Highly effective at reducing vaso-occlusive crises (VOCs). | Base editing avoids double-strand breaks, lowering the risk of DNA damage and gene rearrangements. • Induces exceptionally high fetal hemoglobin levels (averaging >60% in clinical trials). |
| Cons | Double-strand breaks carry theoretical risks of off-target edits, mosaicism, or chromosomal rearrangements. • Long, multi-step treatment process. | Investigational therapy (prepping for FDA approval). • Requires the same rigorous myeloablative conditioning as CASGEVY. |
Molecular mechanism of CASGEVY (CRISPR-Cas9 treatment)
Click here for a video overview of the therapeutic procedure outlined below, and here for a video interview with the first person to be treated with this therapy (Victoria Gray) along with the scientist (Jennifer Doudna) who received the Nobel Prize for her team’s work developing the CRISPR-Cas9 system as recreated in the CRISPR exercise. An overview of this system is provided in Part 1 of the ATTR exercise.
- Hematopoietic Stem Cell (HSC) Extraction:
The patient’s bone marrow stem cells are harvested via apheresis, after inducing movement of the cells from the bone marrow to the blood. - Targeted Gene Editing:
- A guide RNA (gRNA) directs Cas9 nuclease to the erythroid-specific enhancer of BCL11A, as shown in the image below (modified from Fig.1 of Frangoul et al. 2021).
- This enhancer is a crucial regulatory DNA sequence located within the second intron of the BCL11A gene. It controls the expression of BCL11A specifically in red blood cell precursors (erythroid cells).
- Cas9 introduces a double-stranded break at the location of the Cas9 cut site below.
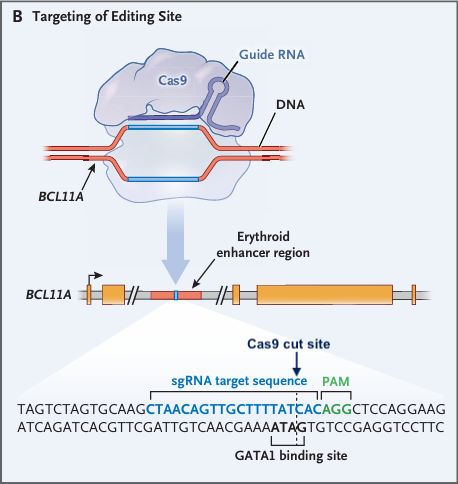
- Disruption of the erythroid-specific enhancer of BCL11A:
- DNA repair occurs primarily via Non-Homologous End Joining (NHEJ), producing insertions or deletions that disable the enhancer. Once the enhancer is disabled, the enhancer-dependent chromatin rosette collapses.
- Functionally, this collapse reduces BCL11A expression in red blood cell precursors, stopping repression of γ-globin genes.
- DNA repair occurs primarily via Non-Homologous End Joining (NHEJ), producing insertions or deletions that disable the enhancer. Once the enhancer is disabled, the enhancer-dependent chromatin rosette collapses.
- Reactivation of Fetal Hemoglobin (HbF):
- The Hematopoietic Stem Cells (HSCs), now engineered to reduce BCL11A activity, preferentially express γ-globin thus increasing HbF production.
- HbF compensates for defective the β-globin in adult hemoglobin, preventing red blood cell sickling.
- The Hematopoietic Stem Cells (HSCs), now engineered to reduce BCL11A activity, preferentially express γ-globin thus increasing HbF production.
- Reinfusion and Engraftment:
- Edited HSCs are transplanted back into the patient after myeloablative conditioning, which clears space in the bone marrow. Note: This procedure has serious side effects but its use is considered justified due to the severe nature of sickle-cell disease.
- The cells engraft and generate a lifelong supply of red blood cells expressing non-sickling HbF.
- Edited HSCs are transplanted back into the patient after myeloablative conditioning, which clears space in the bone marrow. Note: This procedure has serious side effects but its use is considered justified due to the severe nature of sickle-cell disease.
Molecular Outcomes
- HbF competes with HbS, inhibiting polymerization of defective β-globin
- Red blood cells regain flexibility and normal morphology, reducing anemia and preventing vaso-occlusion.
- Patients experience long-term symptomatic relief with a functional cure.
Key Molecular Insights
- CRISPR exploits the cell’s native DNA repair machinery to disrupt a regulatory gene rather than directly correcting the structural mutation.
- Targeting the erythroid-specific enhancer allows selective modulation of BCL11A without compromising its roles in other tissues.
- The treatment demonstrates precision gene regulation, effectively “reprogramming” the patient’s hematopoietic system.
References from Current Research
- CASGEVY (exagamglogene autotemcel) was approved by the FDA in December 2023 using this strategy.
- Clinical trials show high efficacy: most patients maintain HbF levels sufficient to prevent vaso-occlusive crises for extended periods (≥12 months).
- Long-term monitoring is ongoing for potential off-target effects, efficiency of engraftment, and durability of HbF expression.
- The search for an alternative to the current form of myeloablative conditioning continues.
Summary of CRISPR-Cas9 (CASGEVY) treatment
At a molecular level, CRISPR treatment for sickle cell disease reactivates fetal hemoglobin by selectively disabling the erythroid-specific enhancer, using patient-derived stem cells edited ex vivo. This precision-editing restores functional hemoglobin, prevents sickling, and represents a paradigm shift from symptomatic management to curative, gene-level treatment.
Molecular Mechanism of Risto-cell (Base editing treatment)
Watch this video for an animation of the molecular mechanism described below.
- Hematopoietic Stem Cell (HSC) Extraction:
The patient’s bone marrow stem cells are harvested via apheresis, after inducing movement of the cells from the bone marrow to the blood. - Targeted Gene Editing
- A guide RNA (gRNA) directs Cas9 nickase to the promoter regions of HBG1 and HBG2, as shown below (from Fig.1 of Gupta et al. 2026).
Note: HBG1 and HBG2 are two closely related genes that code for different types of gamma-globin proteins. When combined, these proteins are essential building blocks for fetal hemoglobin (HbF). HBG1 produces the A-gamma globin protein. This protein differs from the HBG2 product at a single amino acid, containing an alanine residue at position 136. HBG2 produces the G-gamma globin protein. It contains a glycine residue at position 136, and makes up about 70% of the total gamma globin before birth.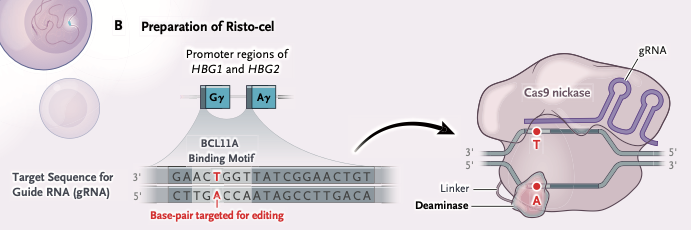
- As shown above, BCL11A binds to a specific distal motif within the promoter regions of HBG1 and HBG2 during the fetal-to-adult developmental switch. This binding recruits corepressor complexes (like NuRD) and hinders the binding of activating factors, such as NF-Y. This base editing technique targets the T-A base pair shown in red above, and results in replacement with a C-G base pair, as shown below (from Fig.1 of Gupta et al. 2026)
- The conversion process involves deamination (removal of an amino group) from adenine, which converts it to inosine. (Note: Inosine is actually hypoxanthine attached to ribose, so technically the term “hypoxanthine” should be used in place of “inosine”. For historical reasons, the shorthand expression that adenine is converted to Inosine is universally used).
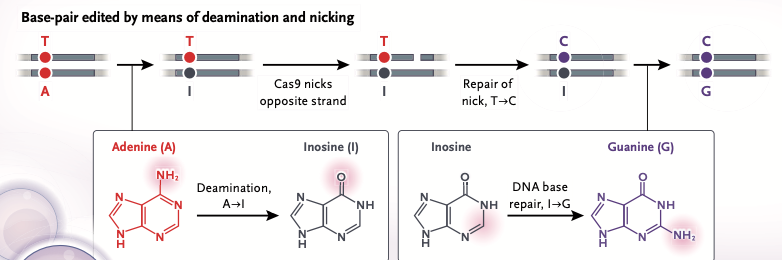
- Cas9 nickase* selectively cuts (nicks) the non-edited, thymine-containing strand. The cell’s mismatch repair machinery degrades the nicked strand but leaves the opposite strand intact.
*The type used was Cas9-D10A nickase, so named because the 10th amino acid in the Cas9 protein chain (Aspartic Acid, symbol D) is mutated into an Alanine (A). Because of this mutation, one of the two cutting domains of Cas9 is deactivated as depicted graphically above (bottom strand is not cut). This form of Cas9 nickase is also used in Part 2 of the CRISPR exercise, recreating research that resulted in the 2020 Nobel Prize in Chemistry (Jinek et al. 2012). - DNA polymerase refills the gap. Because hypoxanthine structurally mimics guanine’s hydrogen-bonding pattern, DNA polymerase preferentially inserts a cytosine opposite the hypoxanthine base. This converts the intermediate mismatch into a stable Hypoxanthine-Cytosine pair.
- The DNA strands separate, with the strand containing the hypoxanthine base seving as a template.
DNA polymerase reads the hypoxanthine base. It presents a keto group at position 6 and a hydrogen at position 1, perfectly matching the hydrogen-bonding donors and acceptors of cytosine. - When the complementary strand is synthesized using this new cytosine-containing template, DNA polymerase inserts a standard guanine opposite the cytosine. The end result is a permanent G-C base pair substitution.
- A guide RNA (gRNA) directs Cas9 nickase to the promoter regions of HBG1 and HBG2, as shown below (from Fig.1 of Gupta et al. 2026).
- Reactivation of Fetal Hemoglobin (HbF):
- The Hematopoietic Stem Cells (HSCs) have been engineered to continue production of γ-globin thus enabling continued HbF production.
- HbF compensates for defective the β-globin in adult hemoglobin, preventing red blood cell sickling.
- The Hematopoietic Stem Cells (HSCs) have been engineered to continue production of γ-globin thus enabling continued HbF production.
- Reinfusion and Engraftment:
- Edited HSCs are transplanted back into the patient after myeloablative conditioning, which clears space in the bone marrow. Note: This procedure has serious side effects but its use is considered justified due to the severe nature of sickle-cell disease.
- The cells engraft and generate a lifelong supply of red blood cells expressing non-sickling HbF.
- Edited HSCs are transplanted back into the patient after myeloablative conditioning, which clears space in the bone marrow. Note: This procedure has serious side effects but its use is considered justified due to the severe nature of sickle-cell disease.
Molecular Outcomes
- As in CASGEVY therapy, HbF competes with HbS, inhibiting polymerization of defective β-globin. Edited stem cells pump out enough HbF so that it makes up the majority of the patient’s hemoglobin, functionally diluting the defective adult HbS and stopping red blood cells from sickling.
- Red blood cells regain flexibility and normal morphology, reducing anemia and preventing vaso-occlusion.
- Patients experience long-term symptomatic relief with a functional cure.
Key Molecular Insights
- Editing of a single base pair is enough to prevent BCL11A from suppressing fetal hemoglobin production.
- Targeting the gamma-globin genes prevents BCL11A from binding but does not suppress the production of BCL11A.
- Like CASGEVY, base editing demonstrates precision gene regulation, effectively “reprogramming” the patient’s hematopoietic system.
References from Current Research.
- Unlike CASGEVY, risto-cell has not yet been approved for clinical use.
- Clinical trials show high efficacy: participants in clinical trials maintain HbF levels sufficient to prevent vaso-occlusive crises for extended periods (≥12 months).
- Like CASGEVY, myeloablative conditioning is required, so a search for an alternative to the current form of this conditioning continues.
Summary of base editing (Risto-cell) treatment
At a molecular level, base editing is used to change a single base pair in the promoters for the gamma globin genes (HBG2 asnd HBG2), using patient-derived stem cells edited ex vivo. This precision-editing restores functional hemoglobin, prevents sickling, and represents a paradigm shift from symptomatic management to curative, gene-level treatment as summarized below (from Fig.1 of Gupta et al. 2026).
Before treatment: Suppression of fetal hemoglobin (HbF) results in HbS polymerization and sickling.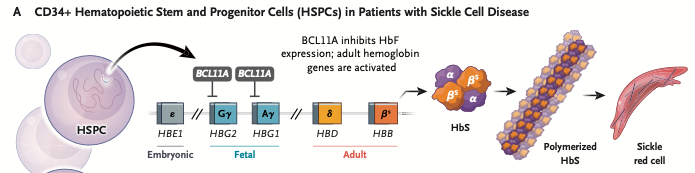
After treatment: Sufficient HbF is produced to prevent HbS polymerization.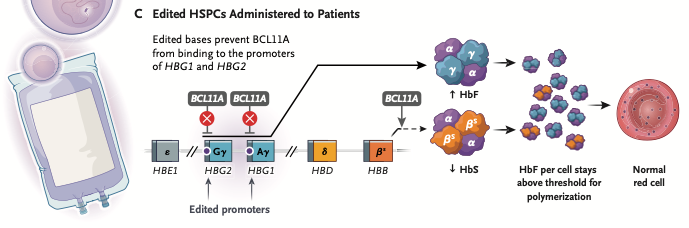
Part 1: Overview of CRISPR-Cas9 and base editing treatments for Sickle cell disease (this page).
Part 2: CRISPR-Cas9 exercise. Use Case It v705 to locate and cut the BCL11A erythroid-specific enhancer, then use BLAST and the Genome Data Viewer to visualize the enhancer.
Part 3: Base editing exercise. Use Case It v705 to locate and edit BCL11A binding sites on the HBG1 and HBG2 promoters, then use BLAST and the Genome Data Viewer to identify and visualize binding sites in relation to gene location.